It's not over until it's all done! There was a major body of work left unfinished which holds pharmaceutical promise to this day and onward.
The Effect of the Human Peptide GHK on Gene Expression Relevant to Nervous System Function and Cognitive Decline.
Quite the noodle baker this evening. Having scoured the literature for literally 10 years regarding Pauling's pivotal ascorbate (uh-skorbate or uhsorbate, tomaytoe, tomahto, ascent, abscond, whatever) work, I somehow developed a blind spot for GHK-copper2+ work. It is incredibly ripe for development.
http://skinbiology.com/copper-peptides-cancer-metastasis-suppression.html
(These are links mind you, not copy-pastes. All copyrights reserved to their corresponding owners.)
Friday, October 20, 2017
Tuesday, August 8, 2017
All roads lead to Rome
No Mevalonate = No new liver cells. Why would I make this up. Too bizarre for me to imagine out of nothingness.
Mevalonate-derived proteins in liver regeneration
The higher end of dosing for statins is ill-conceived. It doesn't do anything other than take a sledgehammer to LDL cholesterol. Lp(a) is still happily made by the liver at the same level, and along with it, Lp(a)'s cholesterol inside it.
That is not the way statins work anyways. When they do work, taken at low doses, it is through non-mevalonate pathway pleiotropic effects discussed in the literature.
The early generation "dirty" statins were so powerful at jamming cholesterol synthesis that they were studied as chemotherapeutics because cells do not divide if they are underneath a threshold of intracellular cholesterol. This body of research is older but easily found.
Mevalonate-derived proteins in liver regeneration
The higher end of dosing for statins is ill-conceived. It doesn't do anything other than take a sledgehammer to LDL cholesterol. Lp(a) is still happily made by the liver at the same level, and along with it, Lp(a)'s cholesterol inside it.
That is not the way statins work anyways. When they do work, taken at low doses, it is through non-mevalonate pathway pleiotropic effects discussed in the literature.
The early generation "dirty" statins were so powerful at jamming cholesterol synthesis that they were studied as chemotherapeutics because cells do not divide if they are underneath a threshold of intracellular cholesterol. This body of research is older but easily found.
Sunday, August 6, 2017
Interleukin-6, the gambit of the crusaders of cholesterol as a plumbing gunk.
As if in possession of a powerful secret bit of information, the petty crusaders of the idea that LDL particles gum up their pathway in the human body, rather than being present at sites of chronic inflammation, will point to IL-6 and snicker as if I didn't know.
IL-6 causes LDL receptor upregulation, and fit and trim appearances.....But also a myriad of degenerative diseases when it is emitted by the immune cells or arterial cells.
One acknowledgement: Atherosclerosis occurs elsewhere besides the coronary arterial bed, the carotids, the cerebrovasculature, the renal bifurcation, and the iliac bifurcations. Because of the mechanical nature of the heart, these plaques are most often lethal there. DeBakey published in detail about this. In fact, it is NOT the heart that most commonly suffers atherosclerosis, but the split that feeds our left and right legs. Yet, this is in line with what the "backwards conspiracy theorists" claim - that atherosclerosis occurs at bifurcations and curves with hemodynamic alteration. There is nothing contrary. Yet, the iliac plaques are generally not lethal. The second most common areas is....the heart arteries.
Patterns of atherosclerosis and their surgical significance.
Yes, IL-6 like nearly all things in the body has a positive function. You need IL-6 signaling.
Inhibiting Interleukin-6 (IL-6): The Key To Health, Successful Aging and Vitality
Some would have a myopic focus on hyper IL-6 reducing plasma lipids and quietly chuckle internally while saying to themselves, "Cholesterol causes heart disease, not IL-6." Not so. Maybe for a while, but the factual mechanism is that chronic IL-6 is destructive and involved in several chronic degenerative diseases, including atherosclerosis. Nothing backwards about anything that anyone has seen. It is a matter of time scale. Short term versus long term. For example, from the link above, "In people who exercised for 3-3.5 hours (marathon exercise), IL-6 increased from 1.5 pg/ml to 94.4 pg/ml immediately post-exercise and to 22.1 pg/ml 2 hours post-exercise (half-life of 1-2 hours) (R, R2). This means blood levels should be completely normal the next day – even after running a marathon." If you had some simmering internal damage emitting the amount of IL-6 generated after a marathon everyday for many years, say a lot of widespread endothelial denudations (torn off lining of the artery), that situation is very different.
In the context of chronic diabetes mellitus, IL-6 drives higher LDL levels in the blood, not lower levels as would be seen in certain acute inflammatory episodes.
Interleukin-6 mediates hepatic hypersecretion of apolipoprotein B
You can have low LDL but high Lp(a). IL-6 is a straightforward upregulator of Lp(a) if not LDL. Lowering IL-6 lowers Lp(a), much more atherogenic than LDL will ever be.
IL-6 blockade by monoclonal antibodies inhibits apolipoprotein (a) expression and lipoprotein (a) synthesis in humans
It doesn't report "IL-6 blockade RAISES blood Lp(a) in humans." Now that would be backwards.
IL-6 causes LDL receptor upregulation, and fit and trim appearances.....But also a myriad of degenerative diseases when it is emitted by the immune cells or arterial cells.
One acknowledgement: Atherosclerosis occurs elsewhere besides the coronary arterial bed, the carotids, the cerebrovasculature, the renal bifurcation, and the iliac bifurcations. Because of the mechanical nature of the heart, these plaques are most often lethal there. DeBakey published in detail about this. In fact, it is NOT the heart that most commonly suffers atherosclerosis, but the split that feeds our left and right legs. Yet, this is in line with what the "backwards conspiracy theorists" claim - that atherosclerosis occurs at bifurcations and curves with hemodynamic alteration. There is nothing contrary. Yet, the iliac plaques are generally not lethal. The second most common areas is....the heart arteries.
Patterns of atherosclerosis and their surgical significance.
Yes, IL-6 like nearly all things in the body has a positive function. You need IL-6 signaling.
Inhibiting Interleukin-6 (IL-6): The Key To Health, Successful Aging and Vitality
Some would have a myopic focus on hyper IL-6 reducing plasma lipids and quietly chuckle internally while saying to themselves, "Cholesterol causes heart disease, not IL-6." Not so. Maybe for a while, but the factual mechanism is that chronic IL-6 is destructive and involved in several chronic degenerative diseases, including atherosclerosis. Nothing backwards about anything that anyone has seen. It is a matter of time scale. Short term versus long term. For example, from the link above, "In people who exercised for 3-3.5 hours (marathon exercise), IL-6 increased from 1.5 pg/ml to 94.4 pg/ml immediately post-exercise and to 22.1 pg/ml 2 hours post-exercise (half-life of 1-2 hours) (R, R2). This means blood levels should be completely normal the next day – even after running a marathon." If you had some simmering internal damage emitting the amount of IL-6 generated after a marathon everyday for many years, say a lot of widespread endothelial denudations (torn off lining of the artery), that situation is very different.
In the context of chronic diabetes mellitus, IL-6 drives higher LDL levels in the blood, not lower levels as would be seen in certain acute inflammatory episodes.
Interleukin-6 mediates hepatic hypersecretion of apolipoprotein B
You can have low LDL but high Lp(a). IL-6 is a straightforward upregulator of Lp(a) if not LDL. Lowering IL-6 lowers Lp(a), much more atherogenic than LDL will ever be.
IL-6 blockade by monoclonal antibodies inhibits apolipoprotein (a) expression and lipoprotein (a) synthesis in humans
It doesn't report "IL-6 blockade RAISES blood Lp(a) in humans." Now that would be backwards.
Tuesday, August 1, 2017
Cholesterol DOES or DOES NOT cause heart disease? Which is backwards?
Given that diabetes causes atherosclerosis, and that cholesterol is never found alone in food, but often with culprits of atherosclerosis, or in a toxic oxidized form, it is easy to confuse the idea that lipoprotein cholesterol DOES NOT cause atherosclerosis with the idea that ox-cholesterol is inflammatory.
If cholesterol were the cause of atherosclerosis, chimpanzees, our nearest genetic cousins in the wild, would all die of atherosclerosis. The fact in the real world is that they do not despite having what is considered extremely high blood cholesterol. Chimpanzees have high cholesterol throughout their entire lives and it gets higher in old age from menopause and andropause. LDL above 170mg/dL, and total cholesterol above 200mg/dL are routine in chimpanzees. They die of heart tissue fibrosis and/or arrythmia resulting from it, but not "clogged arteries."
So, for those who a priori subscribe to the old "cholesterol causes atherosclerosis in people" idea, here is an eye opening look at the facts as they really are.
Heart disease is common in humans and chimpanzees, but is caused by different pathological processes
"This difficulty in inducing advanced lesions in rodent and rabbit models is in stark contrast to the prevalence of severe atherosclerosis in humans, which occurs even in some individuals with mildly elevated LDL levels and no other obvious risk factors (Vasan et al. 2005). Indeed, despite statin treatment of their hypercholesterolemia 70% of such patients still have adverse cardiovascular events (Steinberg 2008)."
"While captive great apes have a relatively healthy diet from the perspective of atherosclerotic risk (low in cholesterol and saturated fat), this is not reflected in their serum lipid profiles. At the Yerkes Primate Center, a mean total cholesterol level of 211.13 ± 38.85 mg/dL was found in a population of 106 male captive chimpanzees over their 50-plus-year lifespan (Herndon and Tigges 2001). Similar ranges were noted at PFA (Howell et al. 2003), and at another center (Hainsey et al. 1993)."
"Second and most intriguingly, we see that unlike the case in humans, captive chimpanzees have high serum cholesterol levels even in the early decades of life (Fig. 2). Thus, the chimpanzee vasculature is exposed to high cholesterol levels throughout adolescence and adulthood. Indeed the levels in very young chimpanzees fall into a high enough range wherein pharmacological statin therapy would now be seriously considered in children (Daniels and Greer 2008)."
The important key fact is the carefully examined result that in humans, after lowering LDL drastically by statins, 70% of these treated patients have CVD events anyways. By simple deductive logic with empirical proof, cholesterol could NOT be the cause of atherosclerosis in these patients with therapeutically lowered cholesterol. So what is the cause? There is the vitamin C deficiency debacle which the deep white hat state knows with certainty to be a major culprit. There are dozens of MOA's for vitamin C in the human cardiovascular and coagulation/hemostasis system. The simple solution is just take lots of vitamin C (and K2, B-complex, and magnesium) for this group and be on with their great noble lives. For the evil hats, it is all about stubborn denial, and mass murdering people around the world indirectly for profit and weird, petty, ulterior motives.
Atherosclerosis happens a lot in people. It is associated with higher LDL levels, but it is NOT cholesterol causing the atherosclerosis. It is there with the atherosclerosis. Hypertension, arterial and blood inflammation, and accidental immune system attacks on the artery wall, mechanical damage, and....insulin resistance and/or diabetes mellitus are increasingly being revealed as the underlying reason FOR high blood cholesterol. We are not chimpanzees, there is a major difference in the aspects of our cardiovascular diseases. The 1% genetic difference is a vast one. Diet in chimpanzees is unrelated to cholesterol which is always high in them regardless of an immaculate vegetarian diet. Diet may however, actually account for their lack of atherosclerosis. The human diet, westernized with globs of trans-fatty acids, rancid oxidized toxic lipids, tons of excess sugar, salt, and hydrogenated oil (alkanes aka gasoline), and void of ascorbate, b-vitamins, magnesium, and vitamin k2 probably is the key to solving this particular mystery. A wild zoo diet will probably put humans back on track to avoiding atherosclerosis too.
If cholesterol were the cause of atherosclerosis, chimpanzees, our nearest genetic cousins in the wild, would all die of atherosclerosis. The fact in the real world is that they do not despite having what is considered extremely high blood cholesterol. Chimpanzees have high cholesterol throughout their entire lives and it gets higher in old age from menopause and andropause. LDL above 170mg/dL, and total cholesterol above 200mg/dL are routine in chimpanzees. They die of heart tissue fibrosis and/or arrythmia resulting from it, but not "clogged arteries."
So, for those who a priori subscribe to the old "cholesterol causes atherosclerosis in people" idea, here is an eye opening look at the facts as they really are.
Heart disease is common in humans and chimpanzees, but is caused by different pathological processes
"This difficulty in inducing advanced lesions in rodent and rabbit models is in stark contrast to the prevalence of severe atherosclerosis in humans, which occurs even in some individuals with mildly elevated LDL levels and no other obvious risk factors (Vasan et al. 2005). Indeed, despite statin treatment of their hypercholesterolemia 70% of such patients still have adverse cardiovascular events (Steinberg 2008)."
"While captive great apes have a relatively healthy diet from the perspective of atherosclerotic risk (low in cholesterol and saturated fat), this is not reflected in their serum lipid profiles. At the Yerkes Primate Center, a mean total cholesterol level of 211.13 ± 38.85 mg/dL was found in a population of 106 male captive chimpanzees over their 50-plus-year lifespan (Herndon and Tigges 2001). Similar ranges were noted at PFA (Howell et al. 2003), and at another center (Hainsey et al. 1993)."
"Second and most intriguingly, we see that unlike the case in humans, captive chimpanzees have high serum cholesterol levels even in the early decades of life (Fig. 2). Thus, the chimpanzee vasculature is exposed to high cholesterol levels throughout adolescence and adulthood. Indeed the levels in very young chimpanzees fall into a high enough range wherein pharmacological statin therapy would now be seriously considered in children (Daniels and Greer 2008)."
The important key fact is the carefully examined result that in humans, after lowering LDL drastically by statins, 70% of these treated patients have CVD events anyways. By simple deductive logic with empirical proof, cholesterol could NOT be the cause of atherosclerosis in these patients with therapeutically lowered cholesterol. So what is the cause? There is the vitamin C deficiency debacle which the deep white hat state knows with certainty to be a major culprit. There are dozens of MOA's for vitamin C in the human cardiovascular and coagulation/hemostasis system. The simple solution is just take lots of vitamin C (and K2, B-complex, and magnesium) for this group and be on with their great noble lives. For the evil hats, it is all about stubborn denial, and mass murdering people around the world indirectly for profit and weird, petty, ulterior motives.
Atherosclerosis happens a lot in people. It is associated with higher LDL levels, but it is NOT cholesterol causing the atherosclerosis. It is there with the atherosclerosis. Hypertension, arterial and blood inflammation, and accidental immune system attacks on the artery wall, mechanical damage, and....insulin resistance and/or diabetes mellitus are increasingly being revealed as the underlying reason FOR high blood cholesterol. We are not chimpanzees, there is a major difference in the aspects of our cardiovascular diseases. The 1% genetic difference is a vast one. Diet in chimpanzees is unrelated to cholesterol which is always high in them regardless of an immaculate vegetarian diet. Diet may however, actually account for their lack of atherosclerosis. The human diet, westernized with globs of trans-fatty acids, rancid oxidized toxic lipids, tons of excess sugar, salt, and hydrogenated oil (alkanes aka gasoline), and void of ascorbate, b-vitamins, magnesium, and vitamin k2 probably is the key to solving this particular mystery. A wild zoo diet will probably put humans back on track to avoiding atherosclerosis too.
Friday, June 2, 2017
Endogenous human Ouabain: Another controversial fact of life
It's there whether you like it or not.
Endogenous Ouabain: Recent Advances and Controversies.
"
EO and Its Isomers
EO was first identified in human plasma 25 years ago.1,2 Despite confirmation in humans and other mammals with mass spectrometry (MS; Figure; Figures S1–S6 in the online-only Data Supplement), nuclear magnetic resonance, and combined liquid chromatography (LC)–immunology methods,3–6 human EO has remained controversial.7
New analytic studies and related findings should allay skepticism. For
example, employment of multistage MS (MS–MS and MS–MS–MS) to examine the
effects of pregnancy and of central angiotensin (Ang) II infusion on EO
in rat plasma led to the discovery of 2 novel EO isomers.8,9
Isomer 1 has MS–MS and MS–MS–MS product ion spectra indistinguishable
from those of EO, but is slightly more polar than EO; it binds to the
antibody used in our radioimmunoassay. Isomer 2 is slightly less polar
than EO, has a distinct MS–MS–MS spectrum, and cross reacts weakly in
our radioimmunoassay. The primary structural difference(s) between EO
and these isomers may involve the steroid nucleus. Importantly, neither
isomer was previously described or is detectable in commercial (plant)
ouabain.8,9"
Endogenous Ouabain And Diabetic Heart Failure: Damned if you do, damned if you don't
Ouabain is synthesized by the kidney in mammals, perplexing some scientists worldwide but creating a field unto itself for others. What confuses some people is that the dogma was that it was a poison only made by plants. What all this reveals is fundamental and profound in understanding heart failure. Global cardiology often focuses on norepinephrine, phenylephrine, dopamine, angiotensin II or ACE, but not endogenous ouabain. It could be the fact that ouabain is both extremely lethal and also simultaneously critical to sustaining extra-oceanic life. While nature has perfected the tight-rope balance as an automation, "humans" can only begin to monkey around with what is often lethal in their 5-digit hands.
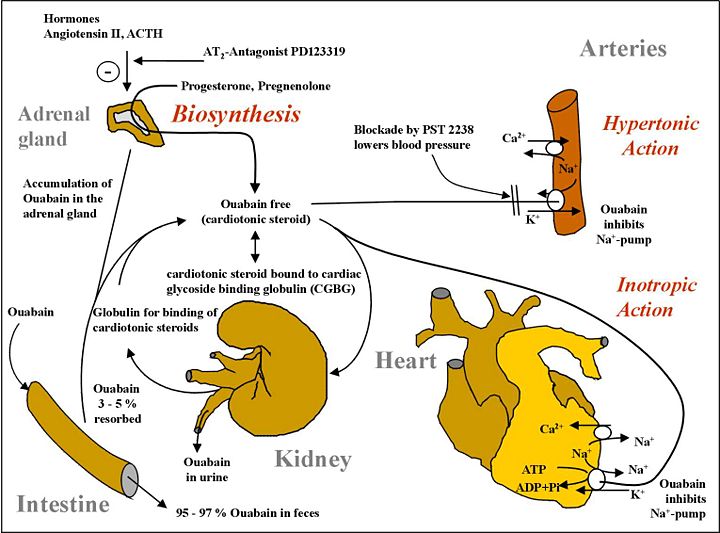
The problem is that with too much ouabain, you overdrive cardiomyocytes to eventual failure, but after renal failure, you do not have enough Endogenous Ouabain (EO), causing decompensated heart failure with structural defects and reduced fractional shortening for a large abnormal diastolic dimension, a weak systolic dimension, and weak ejection fraction. At this point, EO is strangely lower than before it was very high when it caused heart failure through a period of chronic excess. You don't have to use ouabain itself. You may use a similar agonist molecule or even a very different one to stimulate the same calcium overdrive. Of course, this causes problems, but at least it won't be IMMEDIATE death from hypotension, the "dying in your sleep" type of heart failure from thromboses caused by stagnated blood as well as the bulky peripheral resistance of edema. A concomitant problem in diabetic heart failure is no GLUT receptors, therefore no glucose intake, and therefore no ATP synthesis. You can allay this malady with insulinomimetics, many of which have no resemblance to insulin but have potent insulin effects. Polyphenol class molecules can often display impressive insulin receptor pathway stimulation despite having no structural resemblance to insulin. This would re-display glucose receptors for ingestion of glucose into heart cells, restoring the missing ATP synthesis in heart failure. Using insulin is a futile effort as there are no working insulin receptors in diabetic heart failure.
Diabetes mellitus causes dangerous hyponatremia not compensated by endogenous ouabain. Exogenous ouabain rescues diabetic hyponatremia.
The human ouabain synthesis system is to preserve salt at all cost. Sodium chloride, NaCl, is a precious electrolyte, which would be traded for acute and chronic hypertension without reservation via ouabain excretion when salt is available. This is the evolutionary value of NaCl for all creatures that live outside the ocean. Before table salt, sodium chloride availability was sporadic above ocean. This drove our mammalian evolution to conserve salt even if it caused systolic overdrive and hypertension. In the cause of non-diabetes 2 heart failure, AngII or NE and/or ouabain are probably causal. In the cause of IDDM or DM heart failure, the conjoining of reduced ATP synthesis, and renal dysfunction, as well as mildly increased ouabain, suggests that ouabain type agonists could be therapeutic in this scenario to restore cardiac function alone without addressing underlying cardiac insulin resistance as well as renal hormone and electrolyte function impairment.

(above graphic source)
In diabetes, ouabain (sodium conservation) synthesis is deranged:
The sodium pump, an ancestral enzyme with conserved ability to bind ouabain, plays a key role in salt conservation and is regulated by aldosterone and endogenous ouabain (EO). Plasma EO is elevated in about 45% of patients with essential hypertension and correlates with blood pressure. The relationship of EO with Na+ balance is complex. Na+ depletion raises circulating EO, whereas acute saline loads have no effect on EO in essential hypertension, and ambient levels of EO are unrelated to the saline sensitivity of blood pressure. Short-term periods of high dietary salt elevate EO and the relationship with salt balance in normal individuals is V-shaped, whereas the long-term relationship is likely to be L-shaped. Normal individuals suppress the high EO transient triggered by high-salt diets and avoid hypertension. In contrast, patients with elevated EO on normal Na+ intakes have hypertension related to poor modulation of EO biosynthesis, clearance, or both.

The problem is that with too much ouabain, you overdrive cardiomyocytes to eventual failure, but after renal failure, you do not have enough Endogenous Ouabain (EO), causing decompensated heart failure with structural defects and reduced fractional shortening for a large abnormal diastolic dimension, a weak systolic dimension, and weak ejection fraction. At this point, EO is strangely lower than before it was very high when it caused heart failure through a period of chronic excess. You don't have to use ouabain itself. You may use a similar agonist molecule or even a very different one to stimulate the same calcium overdrive. Of course, this causes problems, but at least it won't be IMMEDIATE death from hypotension, the "dying in your sleep" type of heart failure from thromboses caused by stagnated blood as well as the bulky peripheral resistance of edema. A concomitant problem in diabetic heart failure is no GLUT receptors, therefore no glucose intake, and therefore no ATP synthesis. You can allay this malady with insulinomimetics, many of which have no resemblance to insulin but have potent insulin effects. Polyphenol class molecules can often display impressive insulin receptor pathway stimulation despite having no structural resemblance to insulin. This would re-display glucose receptors for ingestion of glucose into heart cells, restoring the missing ATP synthesis in heart failure. Using insulin is a futile effort as there are no working insulin receptors in diabetic heart failure.
Diabetes mellitus causes dangerous hyponatremia not compensated by endogenous ouabain. Exogenous ouabain rescues diabetic hyponatremia.
The human ouabain synthesis system is to preserve salt at all cost. Sodium chloride, NaCl, is a precious electrolyte, which would be traded for acute and chronic hypertension without reservation via ouabain excretion when salt is available. This is the evolutionary value of NaCl for all creatures that live outside the ocean. Before table salt, sodium chloride availability was sporadic above ocean. This drove our mammalian evolution to conserve salt even if it caused systolic overdrive and hypertension. In the cause of non-diabetes 2 heart failure, AngII or NE and/or ouabain are probably causal. In the cause of IDDM or DM heart failure, the conjoining of reduced ATP synthesis, and renal dysfunction, as well as mildly increased ouabain, suggests that ouabain type agonists could be therapeutic in this scenario to restore cardiac function alone without addressing underlying cardiac insulin resistance as well as renal hormone and electrolyte function impairment.

(above graphic source)
In diabetes, ouabain (sodium conservation) synthesis is deranged:
The sodium pump, an ancestral enzyme with conserved ability to bind ouabain, plays a key role in salt conservation and is regulated by aldosterone and endogenous ouabain (EO). Plasma EO is elevated in about 45% of patients with essential hypertension and correlates with blood pressure. The relationship of EO with Na+ balance is complex. Na+ depletion raises circulating EO, whereas acute saline loads have no effect on EO in essential hypertension, and ambient levels of EO are unrelated to the saline sensitivity of blood pressure. Short-term periods of high dietary salt elevate EO and the relationship with salt balance in normal individuals is V-shaped, whereas the long-term relationship is likely to be L-shaped. Normal individuals suppress the high EO transient triggered by high-salt diets and avoid hypertension. In contrast, patients with elevated EO on normal Na+ intakes have hypertension related to poor modulation of EO biosynthesis, clearance, or both.
Saturday, May 27, 2017
Diabetic Hyponatremia
Of course, this is just one problem of many.
Is the association of serum sodium with mortality in patients with type 2 diabetes explained by copeptin or NT-proBNP? (ZODIAC-46).
With chronic hyponatremia, adrenal production of sodium regulating cardiotonic compounds are reduced thereby causing aberrant muscle calcium.
But that is just one problem of several, including a lack of mitochondrial ATP from no glucose import.
Is the association of serum sodium with mortality in patients with type 2 diabetes explained by copeptin or NT-proBNP? (ZODIAC-46).
With chronic hyponatremia, adrenal production of sodium regulating cardiotonic compounds are reduced thereby causing aberrant muscle calcium.
But that is just one problem of several, including a lack of mitochondrial ATP from no glucose import.
Subscribe to:
Comments (Atom)