Given that the guinea pig DOES NOT have Lp(a) but only LDL in a higher ratio to HDL than in wild mice, and that they do incur atherosclerosis upon chronic scurvy, there is no ambiguity that vitamin C deficiency is causal to atherosclerosis WITHOUT invoking apo(a). Unless a genetically modified variant of guinea pig happens, they will never express apo(a) protein. The "immunological evidence" proferred by others is obviously a cross-reaction to a different protein, perhaps even apo(a)-like, but certainly it is not lipoprotein(a). It could not be, rendering any reference to Lp(a) an illogical train of thinking. However, they do have an ample amount of LDL. ApoB particles are important to atherogenesis. We can't just suddenly start ignoring ApoB, not the Lp(a) platform people either. Without ApoB synthesis, amplified by inflammatory arterial cytokines, there is NO Lp(a). In mice that can't make vitamin C but also don't have much LDL but a vast majority of HDL, atherosclerosis is limited to a mild physiological hypertrophy, proteoglycan accumulation, and fibrosis, called arteriosclerosis in olden days. There are some thoracic aortic lesions especially at bifurcations to the digestive system, but little to no atheroma of any visible or clinically significant nature with a majority HDL and few ApoB particles/absolute mass/LDL-cholesterol. This clearly shows that ApoB containing particles outnumbering ApoA containing particles is a precondition to scurvy induced atherosclerosis, as much as scurvy is a precondition to endothelial dysfunction, the cause and initiation of most atherosclerosis. This is without getting into the complicated vasa vasorum component which later contributes to erythrocyte evasation into pre-existing plaques where they die and rupture, releasing membrane cholesterol and iron. (It may be in the very near future, the lipid cores and free cholesterol are found to be from intra-plaque hemorrhage from the interdigitating vasa vasorum rather than the vexing (to some people) foam cell plaque "bust.")
That being said, the atherosclerosis that occurs without Lp(a) is much milder than that which occurs with Lp(a). Atherosclerosis with low Lp(a) and human-esque levels of LDL is much decreased in lesion number, lesion size, and lesion complexity compared to atherosclerosis that occurs with even moderate Lp(a) around. Lp(a) over 30mg/dL certainly will drive more atherosclerosis, but lower levels of Lp(a) below 20mg/dL also contribute by virtue of its "homing missile" like quality of binding to areas of damage which it finds in the system circulation of 60,000 miles. It may not matter how much over a certain threshold of Lp(a), but it does potentiate worse and more with ascending blood levels of Lp(a). There is no question that Lp(a) is atherogenic, but without LDL, how can there be Lp(a)? It is not an "either-or" or "better or worse" proposition, but where all these facts that the 1000's of highly intelligent global cardiology scientists have seen intersect and what that nexus and conjunction of truth is.
Ox-LDL is a reliable marker of vessel disease and number of vessel involvement
Does this mean that Ox-LDL causes atherosclerosis? Yes AND No. It is too low to ever cause atherosclerosis, with circulating levels in multi-vessel CAD being at most 4mg/dL, and normally being 1mg/dL. However, it is an extremely reliable indicator of CAD, with a level of 3mg/dL Ox-LDL most certainly revealing plaque in the coronaries somewhere. Its importance is locally in microenvironments where inflammation causes ROS which causes oxidation and acetylation of LDL in the microscopic milieu around the lesion. If LDL is not oxidized or denatured, it is not "irritating" to the artery, no. Regular LDL, even in very high concentrations has been proven not to be cytotoxic to the endothelial cell or cause stress fiber contraction. As I have already said a long time ago, the LDL gets into the artery if and when the endothelial layer (or vasa vasorum) is compromised and has spatially large enough breaches for it to go through. Vesicular transport would cause LDL to accumulate inside a cell, not outside it, which we'll get to in just a second.
LDL consumption by a cell is highly regulated, and a cell stops internalizing LDL when it has enough, or the cell will turn down its internal synthesis while maintaining internalization. Extracellular aggregates of native LDL could not be there if they could not reach these subendothelial compartments somehow (increased vascular permeability). Ox-and Ac-LDL has a special receptor, scavenger receptor, evolutionarily built in humans, the creature with nearly exclusively the majority of fatal heart attacks and strokes, to get rid of apoptotic cells...and denatured LDL. When scavenger receptor cells encounter ox-LDL, they engulf it without stop. Call it gobbling, but more accurately, they are the "trash men," the "recycling team," not "police men." As much as a policeman can clear debris on a highway, and often does for the public benefit, the macrophage can also serve to clean the artery, not "eat it" or "police" it. The "old concept" people know that fatty streaks, cellular foam cell masses DO NOT burst. So, the cartoon does call out an inaccuracy - the notion that foam cell lesions form, burst open, and evulse their cholesterol into the artery. Early on, no. These are most stable cellular accumulations which would prevent further destabilization of an artery. Later on, if the condition is unresolved, the damage unmended, and the "artery tumor" becomes hypoxic, like in a tumor, the macrophages would undergo necrosis and form a necrotic core, just like happens in a tumor core.
In human plaques, foam cell formation from the smooth muscle cell component is vastly underestimated because in mice, the majority of foam cells occur from macrophages. In humans a greater proportion of foam cells happen from the artery cells themselves. Yet, make no mistake, foam cell "fatty streaks" are found all the time in human atherosclerosis. They may be there, they may not be, but an astute scientist does not simply ignore foam cells, especially those colocalized upon an Lp(a) deposit. There are vastly more human plaques that do not have a foam cell component, and these actually are more worrisome because they are less revertible than a foam cell fatty streak. Why?
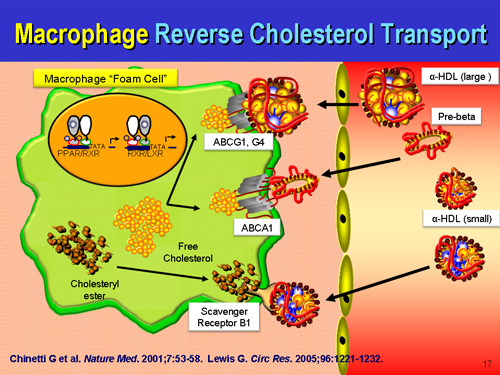
The foam cell, like the apo(a) protein, isn't just some random superfluous curiosity, an accident of biology with no purpose. Foam cells not only contribute to the artery structure temporarily as additional cells with their hydrophobic seal against hemorrhage, cholesterol, but also are participating in transport of excess unused or remaining cholesterol back to the liver, just as LDL delivers useful cholesterol for cell replacement. Of all cell types, the macrophage is very efficient at this process. So we see that this too is a useful, purposeful event, that if resolves satisfactorily, signals the macrophages to exit the artery wall, making the fatty streak lump vanish. Some call it "reverse cholesterol transport," others don't like that name and call it something else. Once again, you can't just ignore foam cells or dismiss fatty streaks. Just as Goldstein and Brown didn't just fabricate a mythic fable of LDL homeostasis, despite this encouraging systemic statin poisoning, scientists and pathologists did not 'just make up' a fable of fatty streaks and foam cells being in human arteries. Truly, these are about repair, rather than a nonsensical auto-attack on the artery wall, although auto-immune arteritis is a real disease too. It is increasingly important to stay updated with the field to see others' understanding of the issue so that all science and scientists everywhere can eventually come to a real-world consensus. Or we can rage around ham-fisted and grandstanding, pretending to know everything without the diligence necessary to know everything.
Macrophage reverse cholesterol transport: key to the regression of atherosclerosis?