Ouabain is synthesized by the kidney in mammals, perplexing some scientists worldwide but creating a field unto itself for others. What confuses some people is that the dogma was that it was a poison only made by plants. What all this reveals is fundamental and profound in understanding heart failure. Global cardiology often focuses on norepinephrine, phenylephrine, dopamine, angiotensin II or ACE, but not endogenous ouabain. It could be the fact that ouabain is both extremely lethal and also simultaneously critical to sustaining extra-oceanic life. While nature has perfected the tight-rope balance as an automation, "humans" can only begin to monkey around with what is often lethal in their 5-digit hands.
The problem is that with too much ouabain, you overdrive cardiomyocytes to eventual failure, but after renal failure, you do not have enough Endogenous Ouabain (EO), causing decompensated heart failure with structural defects and reduced fractional shortening for a large abnormal diastolic dimension, a weak systolic dimension, and weak ejection fraction. At this point, EO is strangely lower than before it was very high when it caused heart failure through a period of chronic excess. You don't have to use ouabain itself. You may use a similar agonist molecule or even a very different one to stimulate the same calcium overdrive. Of course, this causes problems, but at least it won't be IMMEDIATE death from hypotension, the "dying in your sleep" type of heart failure from thromboses caused by stagnated blood as well as the bulky peripheral resistance of edema. A concomitant problem in diabetic heart failure is no GLUT receptors, therefore no glucose intake, and therefore no ATP synthesis. You can allay this malady with insulinomimetics, many of which have no resemblance to insulin but have potent insulin effects. Polyphenol class molecules can often display impressive insulin receptor pathway stimulation despite having no structural resemblance to insulin. This would re-display glucose receptors for ingestion of glucose into heart cells, restoring the missing ATP synthesis in heart failure. Using insulin is a futile effort as there are no working insulin receptors in diabetic heart failure.
Diabetes mellitus causes dangerous hyponatremia not compensated by endogenous ouabain. Exogenous ouabain rescues diabetic hyponatremia.
The human ouabain synthesis system is to preserve salt at all cost. Sodium chloride, NaCl, is a precious electrolyte, which would be traded for acute and chronic hypertension without reservation via ouabain excretion when salt is available. This is the evolutionary value of NaCl for all creatures that live outside the ocean. Before table salt, sodium chloride availability was sporadic above ocean. This drove our mammalian evolution to conserve salt even if it caused systolic overdrive and hypertension. In the cause of non-diabetes 2 heart failure, AngII or NE and/or ouabain are probably causal. In the cause of IDDM or DM heart failure, the conjoining of reduced ATP synthesis, and renal dysfunction, as well as mildly increased ouabain, suggests that ouabain type agonists could be therapeutic in this scenario to restore cardiac function alone without addressing underlying cardiac insulin resistance as well as renal hormone and electrolyte function impairment.
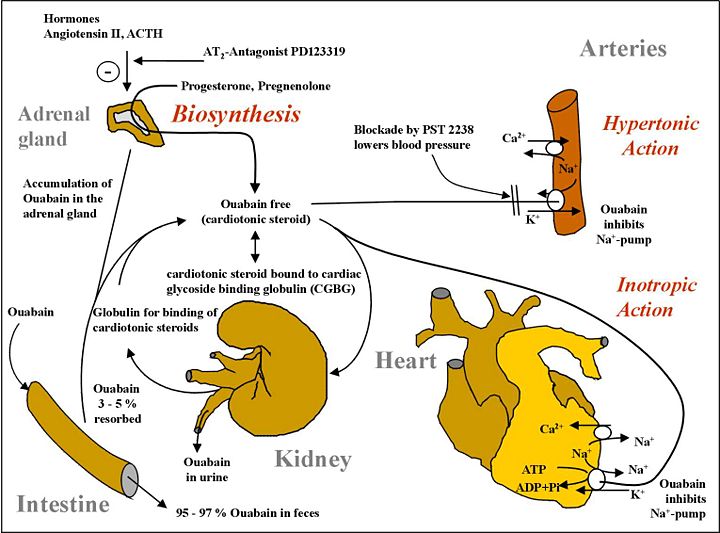
(above graphic source)
In diabetes, ouabain (sodium conservation) synthesis is deranged:
The sodium pump, an ancestral enzyme with conserved ability to bind ouabain, plays a key role in salt conservation and is regulated by aldosterone and endogenous ouabain (EO). Plasma EO is elevated in about 45% of patients with essential hypertension and correlates with blood pressure. The relationship of EO with Na+ balance is complex. Na+ depletion raises circulating EO, whereas acute saline loads have no effect on EO in essential hypertension, and ambient levels of EO are unrelated to the saline sensitivity of blood pressure. Short-term periods of high dietary salt elevate EO and the relationship with salt balance in normal individuals is V-shaped, whereas the long-term relationship is likely to be L-shaped. Normal individuals suppress the high EO transient triggered by high-salt diets and avoid hypertension. In contrast, patients with elevated EO on normal Na+ intakes have hypertension related to poor modulation of EO biosynthesis, clearance, or both.

