The question of causality and time-frame is always to question. The essential point we're trying to make is that Lp(a) doesn't just swoop in from out of nowhere for no reason all the sudden. Macrophages which do home into inflammatory cytokine signals emitted by the artery, and do burrow in to receive cholesterol via active receptor exchange to return to the liver, also don't just suddenly go in and start chewing up lipoproteins. That being said, foam cells and Lp(a) happen together frequently. In any remodeling process in the body, there is nearly certainly found macrophages. You can't just ignore the immune system.
What is my position? It is irrelevant. It is beyond irrelevant "who is right," but what is germaine is "WHAT is right." I do not care "who" is right, nor should you. Those who care intensely about who should be right obviously have something else driving them like a jerk's ego or some frivolous vendetta. The question should be, how do we make each and every one of the millions of people of Earth right in their application of science. What we should all care about is "what is right," not "who is right." Needless to say, that is my concern in science, no extraneous nonsense.
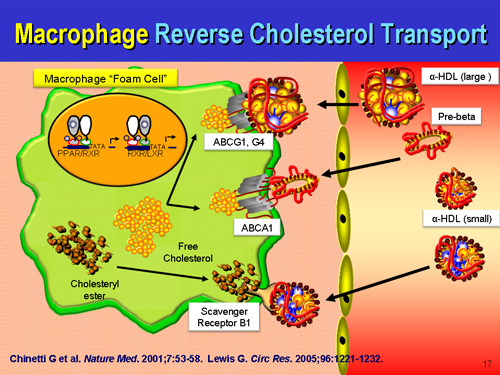
What is true is that macrophages are initially participating in a beneficial remodeling process, and not chewing up ox-LDL in some parasitic process, but attempting to hand back the remodeling waste, the construction site debris back to the liver for recycling via....HDL. We certainly can't ignore HDL in this repair process either. It will spin cardiopathology's noodle when they see that the majority of atherosclerotic plaques are rich in ApoE, which if extended the old logic applied to LDL is causing harmful things. On the contrary, the presence of ApoE we all know to be part of the repair process which may be overwhelmed by a net excess of damage as opposed to a net excess of repair.
Does Lp(a) cause atherosclerosis? Only if there is a reason for atherosclerosis to take place. For a many humans who eat just an orange a day but still have the Gulo-/- genetic defect, there is a lot of cumulative damage that does occur which needs patching by Lp(a) and the coagulation system. Think of it this way, when thinking about preserving arterial integrity: a normal rat makes the equivalent of 5 grams a day in a 70 kilo human when nothing is going on. In stressful times, a rat makes 300% more vitamin C. This entry certainly is not encouraging or supporting the fallacy that dietary cholesterol causes heart disease. It does not, but other dietary factors such as too low vitamin C, B, E, magnesium, K, D, amino acids and too much trans-fatty acids and omega-6 do. The recent US Federal guidance to stop paying attention to ingested cholesterol is a well-studied one made with decades of clinical evidence that cholesterol consumption has historically had nothing to do with the rate of heart disease.
I think this statement I made a long time ago sums it up nicely about what is going on:
"A big lump is better than a big hole."
One prevents lethal hemorrhage, the most dramatic example in human biology being the aortic dissection with adventitial failure, the other is lethal hemorrhage. One of the first priorities of the human physiology is to stop bleeding and hemorrhaging. It drops everything it is doing and attends to that first, even if it means making a pile of disorganized stuff at the site of leaking that is harmful down the road. Better than bleeding to death. Lp(a) is very atherogenic at sites of arterial damage, and sticks more to the glycocalyx with ascending concentrations. If the endothelium is in tact, there is nothing for Lp(a) to react to, no ligand, no binding site to the ligand. Lp(a) just floats along harmlessly as it does not encounter plasminogen binding sites, free lysyls, fibrin, exposed subendothelial fibronectin, etc. You can't just ignore the coagulation system either. Lp(a) has a direct affinity to fibrin(ogen) which is Clotting Factor 1, and gets cross-linked to fibrin clots via FactorXIII. Do macrophages foam cells CAUSE atherosclerosis? No. They are attempting to prevent it, but the process can and does go wrong if they get overwhelmed. When they are participating in arterial repair, foam cells are formally defined as early plaques, but that is simply a matter of scientific semantics. Perhaps one day, they will be relabeled as "reparative cells" distinct from a fibrofatty mass devoid of cells that can rupture and cause thrombosis. Given an equal sized foam cell lesion and fibrofatty mass with thin cap that is less than 20% occlusive, the unstable fibrofatty mass is the dangerous thing, and the foam cell lesion is unconcerning.
Macrophages and foam cells are not always in human atherosclerotic plaques. In fact, many times, they are not there at all. Many pathologist specimens of human diseased atherosclerotic arteries do not have foam cells, which is why some scientists choose to de-emphasize them or diminish their importance. But they do happen, and we can't just ignore them. One improvement in the future state-of-the-art may be to put macrophages and foam cell "lesions" in a category all on their own as they are dynamically different from all other component atherosclerotic plaques, and not just different in cellular origin and composition. They also serve an entirely different purpose altogether than other cells found in human plaques, including remodeling, debris clearance, and returning the artery back to its original state.